
Molecular AI by Empirical Bio
Frontier AI driven biomolecule design for biotech firms
Tools that predict and optimise biomolecule structures
Developed In Association With

AI Research
Building world models at molecular scale
Next-Gen Foundation Models
Our focus is on scaling AI at the intersection of deep learning and physics.


AI Platform
GEMS: Empirical Exploration of Molecular Space
The AI operating system for drug discovery, powered by SOTA models
GEMS: the agentic end-to-end solution
Drug hunters + agents run their full design workflows 24/7 on GEMS
Generate
Supercharging chemical creativity
Empirical chemists use GEMS to assist with molecule ideation, either at massive scale during Hit ID or highly targeted exploration during lead optimization. This accelerates design-make-test cycles by using the power and creativity of our platform to propose new synthesizable ideas for in silico screening.
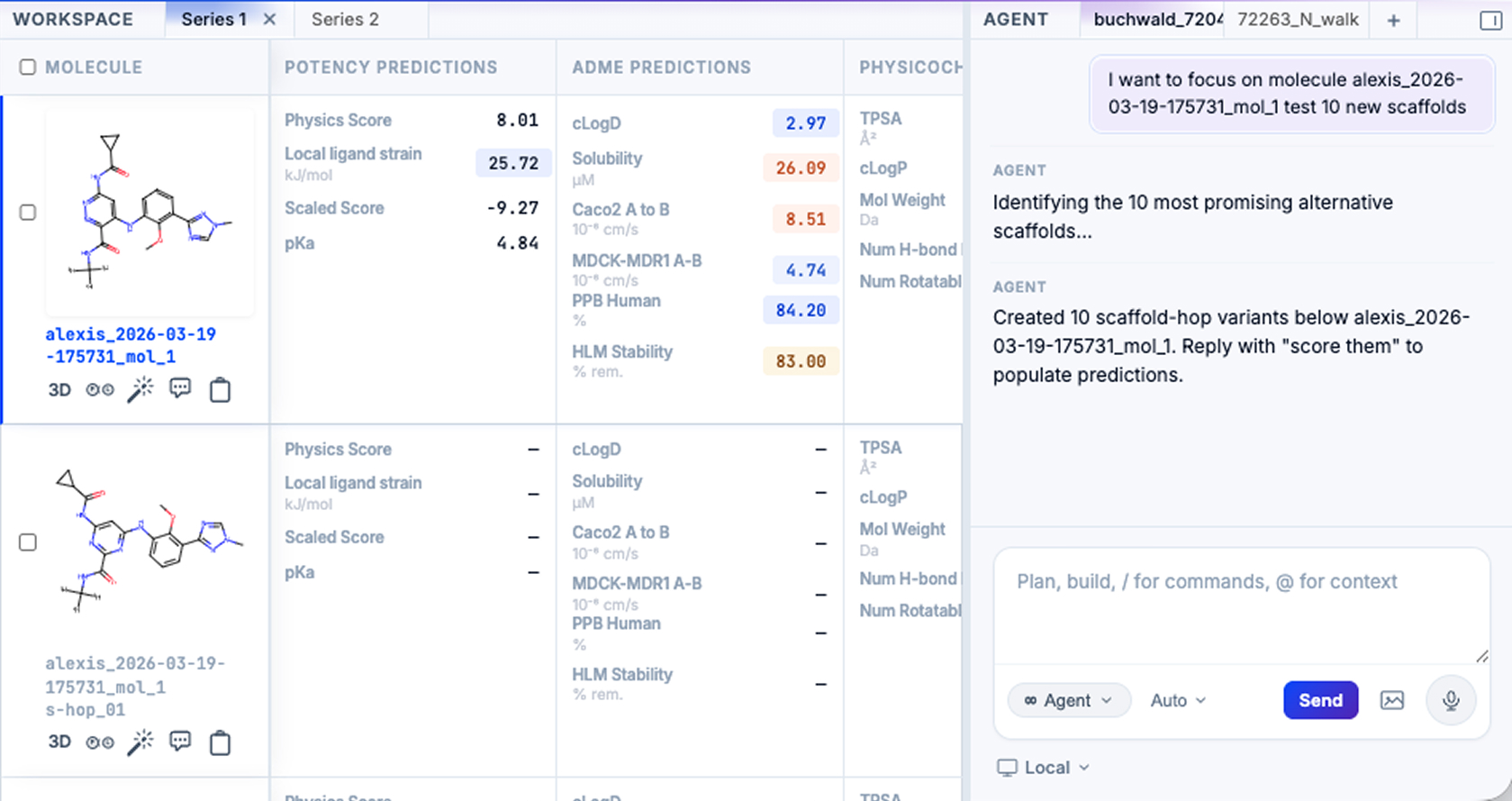
Predict
Predicting structure, potency, and molecular attributes
GEMS uses Pearl, our foundation model, to rapidly and accurately predict 3D structures of protein-ligand complexes, offering insights into binding mechanics and enabling accurate downstream predictions on potency and selectivity. GEMS also predicts a wide range of ADME characteristics using multitask ML models.

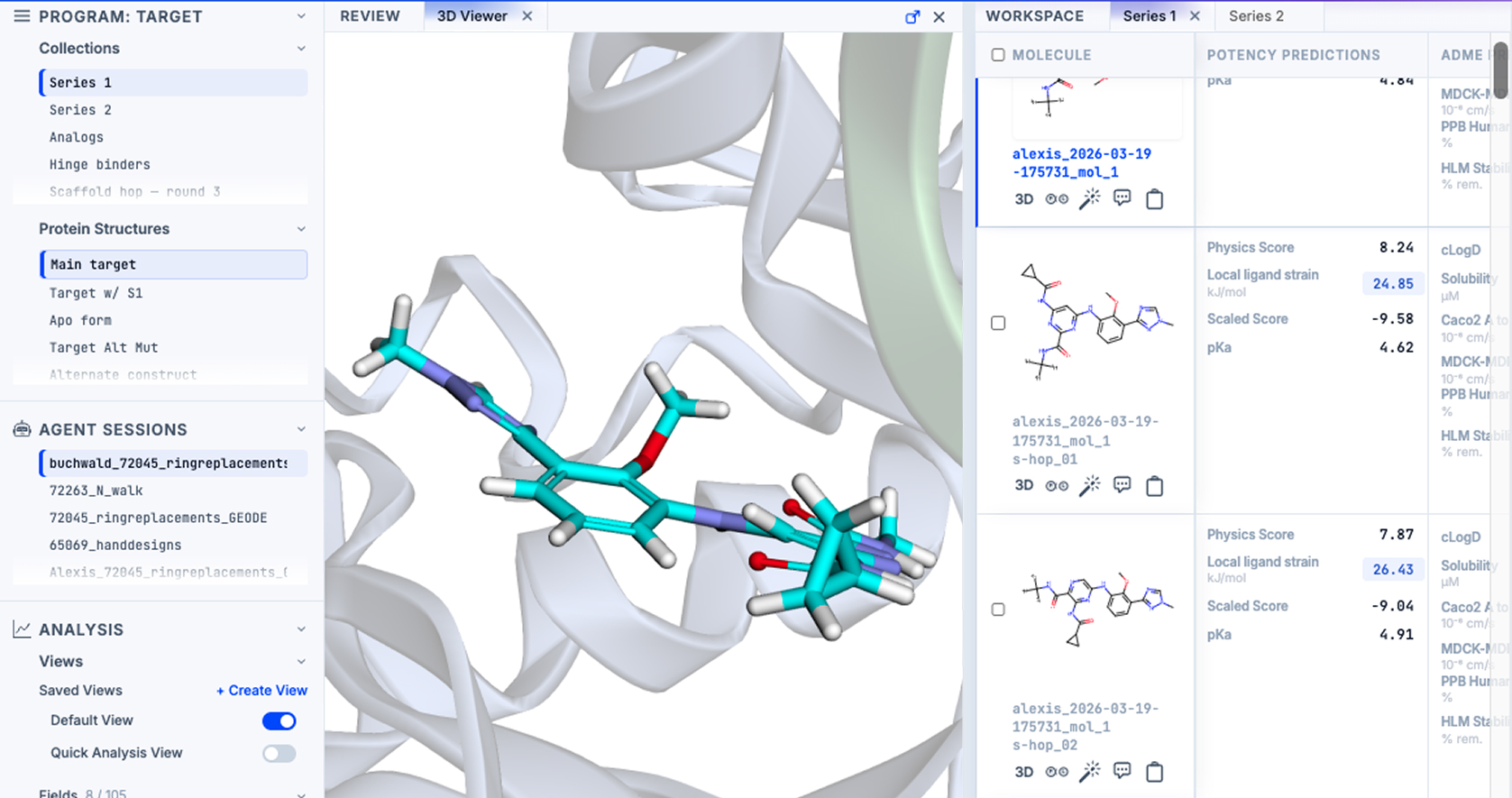
Interrogate
Putting chemists in the driver's seat
GEMS equips chemists with the tools to analyze and interrogate platform predictions. They can visualize 3D structures, deploy agents or filter and sort manually, and compare compounds across the optimization axes that matter for the program.
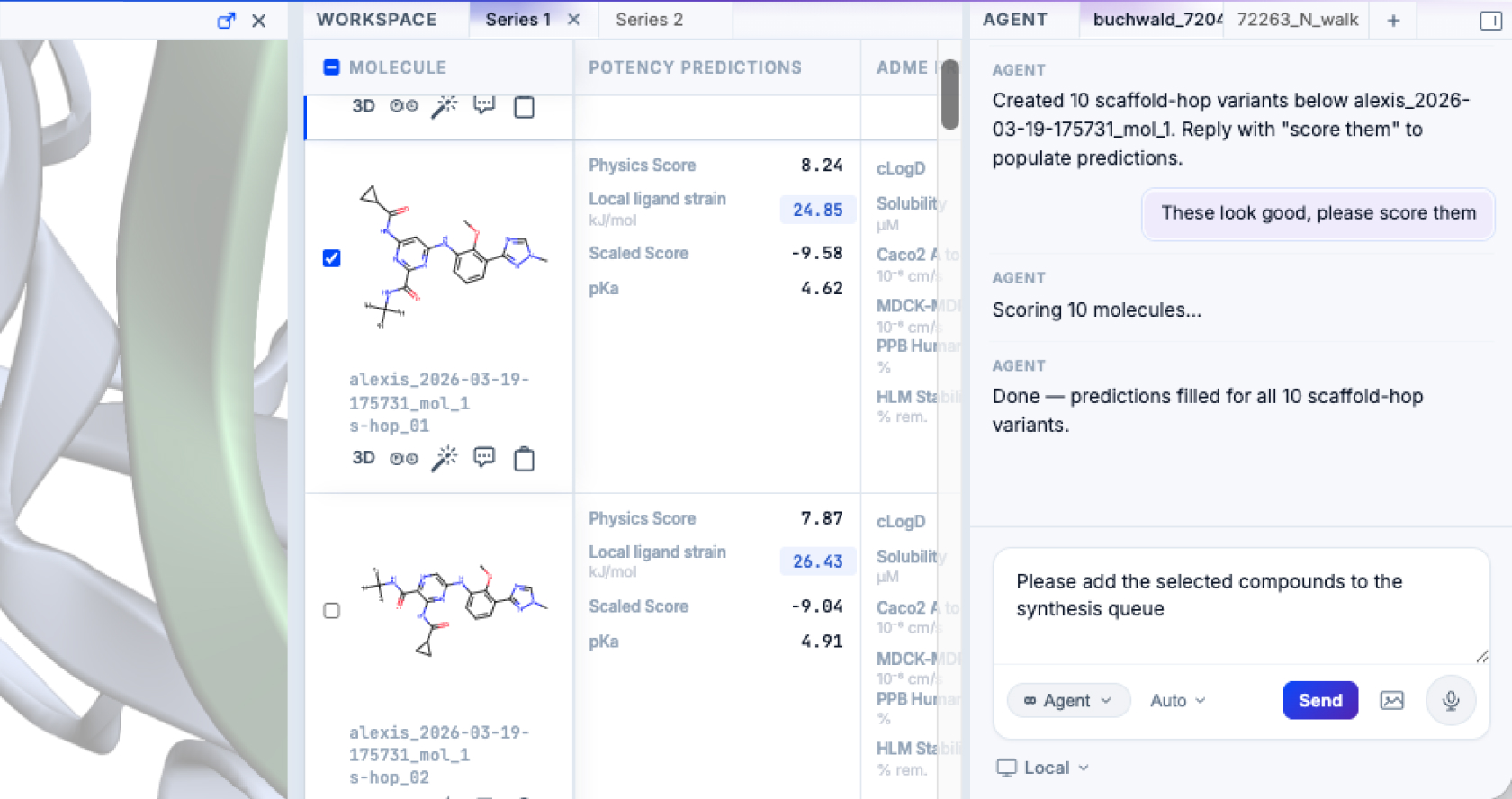
Decide
Better decisions, faster
Chemists move faster from prediction to plan. They use predicted 3D structures to design more potent and selective compounds and can surface trade-offs between molecular characteristics at a glance. The platform streamlines the end-to-end design process.
Platform Intelligence
GEMS is powered by our industry-leading models for small and medium-size molecule drug discovery
Pearl: our industry-leading foundation model for 3D structure prediction
Pearl predicts the 3D structures of protein-ligand complexes at the <1Å RMSD accuracy drug discovery demands. Pearl is trained on physics-based synthetic data proprietary to Empirical, and can be fine-tuned with program-specific data to improve accuracy on individual targets. Chemists can condition Pearl on what they know about their target, steering predictions toward the structures that matter for their program.
Controllable molecular generation
GEMS proposes novel, drug-like, diverse, and synthesizable molecular ideas conditioned on what the chemist is trying to achieve, including ADME, structural, and program-specific constraints. Chemists drive the design strategy, and the platform surfaces candidates worth pursuing.
Potency and selectivity prediction
GEMS integrates structure-based deep learning methods with physical simulation, including molecular dynamics and quantum chemistry, to predict potency and selectivity. This enables Empirical to find drug candidates for challenging targets that lack on-target training data.
ADME property prediction
Using multitask ML models, GEMS predicts 30+ key ADME properties, including solubility, permeability, metabolic stability, and many others. Chemists see signals for drug-likeness on every candidate before deciding what to make.
Controllable molecular generation
Our language models propose novel, drug-like and diverse molecular ideas conditioned on what the chemist is trying to achieve, including ADME, structural, and program-specific constraints. Chemists drive the design strategy, and the platform surfaces candidates worth pursuing.
Potency and selectivity prediction
GEMS integrates structure-based deep learning methods with physical simulation, including molecular dynamics and quantum chemistry, to predict potency and selectivity. This enables Empirical to find drug candidates for challenging targets that lack on-target training data.
ADME prediction
GEMS predicts 30+ key ADME properties using multitask ML models. Chemists see signals for drug-likeness on every candidate before deciding what to make.
Platform flywheel
GEMS is actively shaped by the drug hunters across the industry who use it every day.
